

Jun 06, 2025
Gene Therapy Manufacturing
Gene Therapy Manufacturing Facility: Design, Cleanroom Standards, and Modular Innovation
Introduction: What Is a Gene Therapy Manufacturing Facility?
A gene therapy manufacturing facility is a highly specialized environment where genetic material is produced, packaged, and formulated for therapeutic use in humans. These facilities are responsible for manufacturing viral vectors, plasmids, and gene-edited cell products used to treat genetic disorders, cancers, and rare diseases by delivering corrective or therapeutic genes directly into patient cells. Given the complexity, regulatory sensitivity, and biological risk associated with gene therapy products, these facilities must be engineered to meet the most stringent standards for sterility, biosafety, and product consistency.
Unlike traditional biologics manufacturing, gene therapy production involves smaller batch sizes, high variability, and often patient-specific processing. Cleanroom classification, biosafety containment, and GMP compliance must be tightly integrated into every stage of the facility’s design. Additionally, processes such as upstream vector production, downstream purification, and aseptic fill-finish must be spatially and operationally separated to prevent cross-contamination and maintain product quality.
With the rapid growth of gene therapy pipelines and commercial approvals, modular cleanroom systems have emerged as a vital tool for building these facilities faster and with more flexibility. Modular facilities offer the ability to scale, reconfigure, and validate production space without the delays and costs associated with traditional construction—making them particularly well-suited to this dynamic and fast-evolving field.
This article explores the key components of gene therapy facility design, including cleanroom zoning, ISO and biosafety classifications, GMP requirements, and the advantages of modular cleanroom platforms for both clinical and commercial production.
Core Processes in Gene Therapy Manufacturing
Gene therapy production involves several interdependent stages that must occur in controlled environments to ensure safety, sterility, and efficacy. These stages include upstream vector production, downstream purification, formulation and fill-finish, and extensive quality control testing. Each phase requires purpose-built cleanroom infrastructure and rigorous process validation to meet regulatory expectations for both clinical-grade and commercial products.
Upstream Production
This phase involves the creation of viral vectors—most commonly adeno-associated virus (AAV), lentivirus, or retrovirus—used to deliver therapeutic genes. Vectors are produced using transfected mammalian or insect cells, often in adherent or suspension-based bioreactor systems. This step requires biosafety containment (typically BSL-2) to protect operators from exposure to replication-competent viruses or genetically modified organisms (GMOs).
Downstream Purification
After production, vectors are harvested from the culture media and purified to remove host cell proteins, DNA, and process-related impurities. Common techniques include depth filtration, tangential flow filtration (TFF), chromatography, and ultracentrifugation. These processes require cleanroom conditions that support aseptic control while allowing for flexible, equipment-intensive workflows.
Formulation and Fill-Finish
Purified vectors are formulated with stabilizing buffers and filled into final containers, such as vials or syringes. This stage is performed under ISO 5 conditions using automated filling lines, isolators, or biosafety cabinets, and represents a critical control point for sterility assurance. Final product must meet identity, purity, potency, and sterility specifications before batch release.
In-Process and Quality Control Testing
Throughout manufacturing, extensive analytical testing is required to monitor viral titer, genomic integrity, residual impurities, and sterility. Many gene therapy facilities include adjacent or integrated QC labs with environmental controls and segregation from production zones.
Plasmid DNA Production
Plasmid DNA used in vector generation must also be manufactured under GMP conditions, typically in separate cleanroom suites or offsite facilities. Coordination between plasmid suppliers and vector production teams is essential to avoid delays and ensure material traceability.
Each of these processes introduces unique cleanroom and biosafety requirements. Proper zoning, contamination control, and material/personnel flows are essential to ensure product quality, worker safety, and regulatory compliance throughout the gene therapy manufacturing lifecycle.
Cleanroom Classification and Containment Requirements
Due to the biological nature of gene therapy products, cleanroom environments must meet both ISO particle classification standards and biosafety containment requirements. This dual mandate creates a complex design challenge that must be addressed from the earliest planning phases.
ISO Cleanroom Classifications
- ISO 5 (Grade A): Required at points of open product exposure, such as filling and aseptic connections. Achieved through laminar airflow hoods, isolators, or restricted-access barrier systems (RABS).
- ISO 7 (Grade B/C): Used as background environments for ISO 5 zones and for downstream purification or formulation activities. These areas typically maintain 30+ air changes per hour (ACH) with HEPA filtration and positive pressure.
- ISO 8 (Grade D): Serves as a buffer or support area for gowning, staging, or material entry. These zones are monitored for nonviable particles and may support unclassified storage or prep activities.
Biosafety Level (BSL) Requirements
Gene therapy facilities must also comply with biosafety regulations when working with replication-competent viruses, GMO vectors, or human-derived cell lines. Most operations fall under:
- BSL-2: Required for handling viral vectors such as AAV or lentivirus. Includes Class II BSCs, biosafety training, autoclave access, and restricted facility access.
- BSL-2+ or Enhanced BSL-2: Includes additional controls such as negative pressure rooms, airlocks, and PPE upgrades when working with high-titer or novel vectors.
- BSL-3: Rare in gene therapy, but may be required for more virulent or airborne vectors under development.
Zoning and Containment Strategies
Facility layouts must clearly separate upstream and downstream processes, with airlocks and pressure differentials between zones. Material pass-throughs and gowning rooms are required to enforce unidirectional flow and minimize contamination risks. If both BSL and GMP requirements apply to the same space, the design must harmonize both systems without compromising operator safety or product quality.
Airflow, Filtration, and Pressure Cascades
HEPA filtration is standard in ISO 7 and ISO 5 zones, with directional airflow patterns engineered to reduce turbulence and protect critical zones. Pressure cascades are used to prevent vector escape (in BSL zones) or environmental ingress (in sterile zones), depending on the process and phase.
Complying with both GMP and biosafety standards demands a facility that is robust, flexible, and precisely engineered. Modular cleanrooms provide an ideal platform for integrating these systems while accelerating project timelines and reducing risk.
Facility Layout and Workflow Design for Gene Therapy
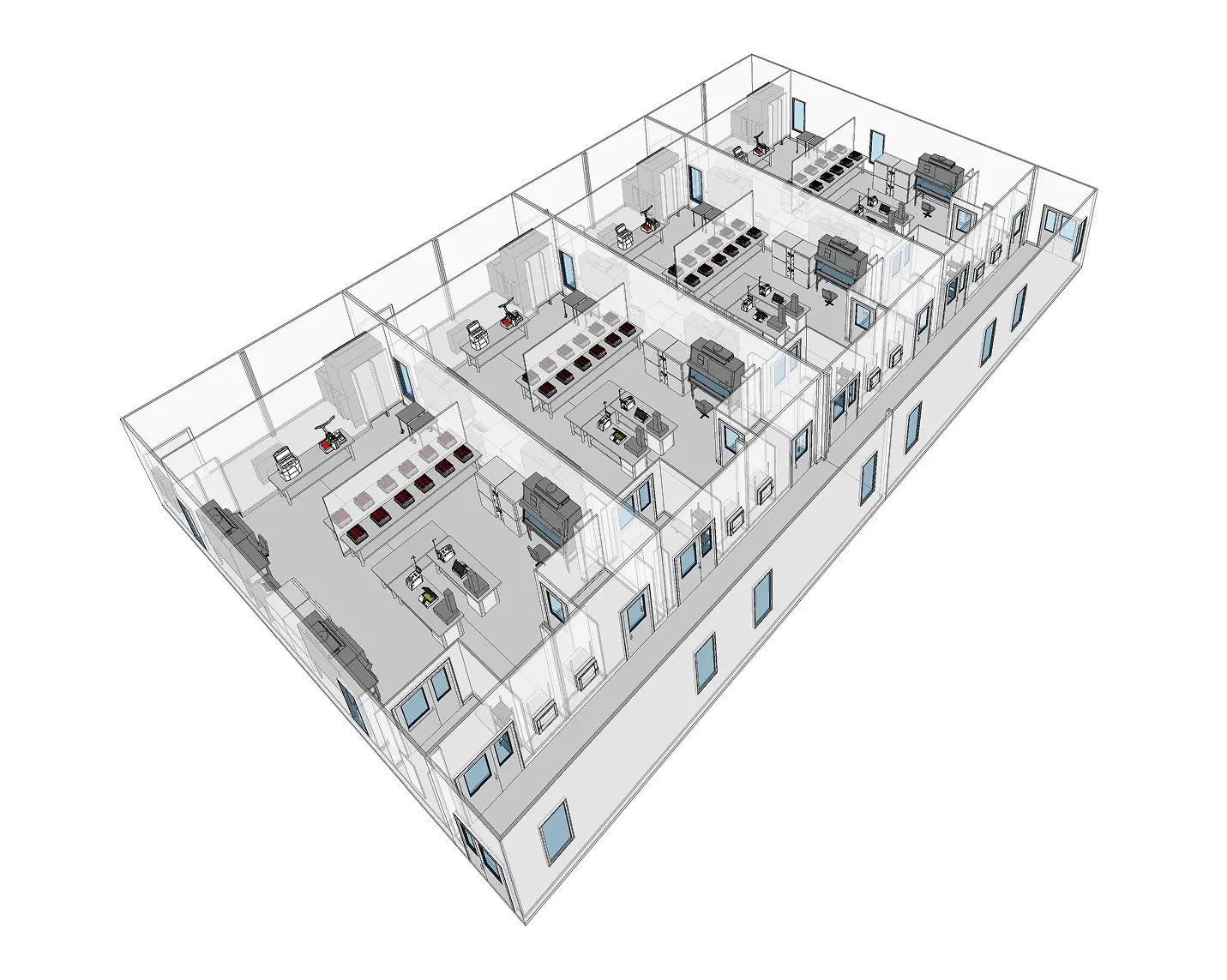
Designing a gene therapy manufacturing facility requires careful attention to spatial layout, room classification, personnel and material flow, and cross-contamination controls. Due to the complexity of gene therapy processes—from upstream vector production to final fill-finish—facilities must be structured to separate incompatible operations, enforce GMP and biosafety zoning, and optimize efficiency within tight physical and regulatory constraints.
Zoned Cleanroom Suites
Facilities are divided into distinct zones for upstream processing, downstream purification, formulation, fill-finish, and storage. Each zone must support the cleanroom classification and containment level required by the process taking place. For example, upstream vector production may require a BSL-2, ISO 7 environment, while fill-finish demands ISO 5 airflow with GMP-grade environmental monitoring.
Unidirectional Personnel and Material Flow
Workflow must follow a forward-moving path from raw materials to finished product, minimizing crossover between clean and dirty areas. Gowning rooms, airlocks, and pass-through chambers help control traffic and support chain-of-identity protocols. Separate entry and exit routes for personnel and materials reduce contamination risks and improve inspection readiness.
Separation of Upstream and Downstream Operations
Upstream processes involve handling high-titer viral vectors and live cell cultures, creating a heightened risk of cross-contamination. These areas must be separated from downstream fill-finish and packaging operations, ideally with independent air handling systems and access controls.
Dedicated Support Areas
Gene therapy manufacturing also requires multiple support zones, including plasmid storage, buffer prep, cell thawing, QC labs, equipment staging, and waste decontamination. These areas must be designed to maintain cleanliness while supporting rapid batch turnover and reliable access to critical materials.
Equipment Integration and Scalability
Facility layouts should support single-use systems, automated bioreactors, chromatography skids, and isolator-based fill lines. Space for future expansion and technology upgrades should be planned from the outset, especially as gene therapy workflows evolve toward closed-system and high-throughput models.
An effective layout streamlines processes, minimizes contamination risk, and maximizes flexibility. The goal is to enable safe, compliant, and efficient production while remaining adaptable to new vectors, platforms, or regulatory shifts.
The Role of Modular Cleanrooms in Gene Therapy Manufacturing
Modular cleanrooms have become a cornerstone of modern gene therapy infrastructure, offering manufacturers the ability to rapidly build, qualify, and scale cleanroom space while maintaining full GMP and biosafety compliance. Whether producing AAV vectors, lentivirus, or genetically modified cells, modular systems provide a proven, accelerated path to facility readiness in a market defined by tight timelines and evolving technologies.
Speed to Deployment
Gene therapy companies cannot afford to wait years for traditional construction. Modular cleanrooms can be fabricated offsite while site prep occurs in parallel, dramatically shortening time to validation and batch release. This speed is critical for clinical-stage companies racing toward pivotal trials or commercial firms launching new indications.
Prevalidated ISO and BSL Compliance
Leading modular providers deliver units prequalified for ISO 5, 7, or 8 classifications and capable of meeting BSL-2 or enhanced biosafety requirements. HEPA filtration, pressure differentials, and airflow patterns are tested during factory acceptance testing (FAT), streamlining on-site commissioning.
Segregated PODs for Containment and Control
Modular PODs can be deployed as isolated suites for upstream production, downstream purification, or final formulation. This compartmentalization supports contamination control, facilitates GMP zoning, and simplifies containment strategies for high-risk vectors or genetically modified organisms.
Built-in Environmental Monitoring and BMS
Modular cleanrooms come equipped with integrated environmental monitoring systems (EMS) and building management systems (BMS) to track airborne particulates, pressure cascades, temperature, and humidity. These systems support 21 CFR Part 11 compliance and provide audit-ready reporting for inspections.
Flexibility and Scalability
As product pipelines evolve or regulatory requirements change, modular systems can be reconfigured or expanded. New PODs can be added without disrupting validated operations, and cleanroom suites can be tailored to accommodate different vector types or production scales.
Reduced Risk and Capital Predictability
Compared to traditional construction, modular cleanrooms reduce design complexity, change orders, and budget overruns. Standardized components, repeatable layouts, and defined validation protocols help reduce both construction risk and long-term operational uncertainty.
In a highly regulated and fast-moving field like gene therapy, modular cleanrooms deliver the infrastructure agility needed to bring innovative therapies to market—faster, safer, and more efficiently.
Future-Ready Infrastructure for Gene Therapy Manufacturing
Gene therapy represents one of the most transformative advances in modern medicine—offering curative potential for genetic diseases, cancers, and previously untreatable conditions. But the success of gene therapy hinges not only on scientific discovery, but on the ability to manufacture these complex products safely, consistently, and at scale.
Gene therapy manufacturing facilities must combine ISO-classified cleanroom environments with biosafety containment, robust zoning, and flexible, GMP-compliant workflows. They must support viral vector production, aseptic fill-finish, chain-of-identity protocols, and real-time monitoring across every step. Traditional construction often fails to meet the speed and adaptability this field demands—making modular cleanrooms an ideal solution.
Modular cleanroom platforms allow gene therapy companies to move faster, reduce capital risk, and meet stringent regulatory standards from clinical development through commercial launch. With prevalidated ISO and BSL zoning, integrated EMS/BMS systems, and scalable layouts, modular facilities are the modern backbone of gene therapy production.
In an industry defined by innovation and urgency, the infrastructure must keep pace. Modular cleanrooms enable gene therapy pioneers to accelerate discovery, streamline compliance, and deliver life-changing treatments to patients around the world.
Frequently Asked Questions About Gene Therapy Manufacturing Facilities
What cleanroom classifications are used in gene therapy facilities?
ISO 5 is required for open aseptic operations like fill-finish. ISO 7 and ISO 8 rooms support upstream production, purification, staging, and gowning areas. Cleanroom zoning is combined with biosafety protocols based on the vector type.
What biosafety level is required for gene therapy?
Most gene therapy processes require BSL-2 containment, especially when working with viral vectors such as AAV or lentivirus. Enhanced BSL-2 (BSL-2+) or BSL-3 may be required for certain high-risk vectors or novel constructs.
Can modular cleanrooms meet both GMP and BSL requirements?
Yes. Modular cleanrooms can be engineered to meet ISO 14644 standards for cleanroom classification and CDC/NIH biosafety level requirements simultaneously, with proper airflow, pressure controls, and containment systems.
How quickly can a modular gene therapy facility be deployed?
Modular cleanroom PODs can be designed, built, installed, and validated in as little as 12-18 months—significantly faster than traditional construction timelines.
Why is segregation important in gene therapy manufacturing?
Segregation prevents cross-contamination between process steps, batch runs, and different vector types. It is essential for maintaining product safety, especially in facilities handling replication-competent or genetically modified materials.